

Abstract
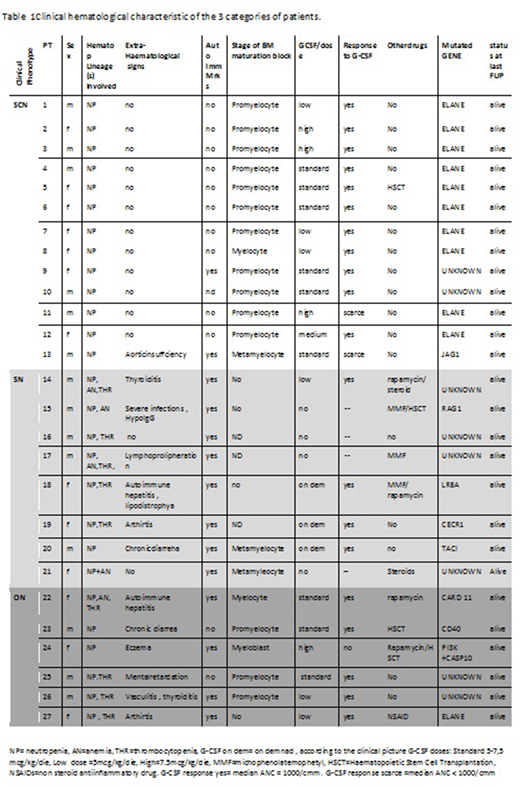
Severe Chronic Neutropenia (SChrN) is an heterogeneous group of disorders characterized by persistently low circulating neutrophils (<500/cmm). Within this group, the classical form of Severe Congenital Neutropenia (SCN), once defined Kostman's disease, appears in early infancy, usually shows a marrow block at promyelocyte stage and needs lifetime treatment with G-CSF to prevent severe infections. Mutations in more than 20 genes have been recognized as causative of the disease with a clear prevalence of ELANEgene mutations (70% of cases). However,SChrN may also be an epiphenomena of autoimmune/immune dysregulation disorders (Secondary Neutropenia, SN) which are usually associated to extrahaematological signs and/or positivity of autoimmunity markers, to a normal or "left shifted" bone marrow morphology. In spite of these categorization many cases do not fit either group and share features of both of them. These "Ovelap Neutropenia" (ON) patients represent a challenge for diagnosis and management. AIM OF the study: Investigate the genetic backgroud of a cohort of SChrN subjects screened in two immune-hematology centers in Italy. Material and Methods: Patients SChrN were prospectively seen in participating Centres and diagnosed/followed-up according to published guidelines (3,4). Genetic diagnosis included classical Sanger technique for commonest severe chronic neutropenia genes and an enlarged NGS panel including also genes responsible for PID. Results: From 2008 to 2016, 27 SChrNpatients (52% males)with a median age at last follow of 13y (range 20 mos-53yrs ) entered the study (Table 1).
Thirteen/27 subjects (48%) were phenotypically diagnosed as classical SCN. Of them 10 (77%) and 1 (8%) had mutations in ELANEand JAG1 genes respectively and 2 (16%) had no pathogenic gene detectable. Eight/27(30%) patients were phenotypically diagnosed as SN and the remaining 6/27 ( 22%) had the clinical features of ON. A pathogenic mutation in a Primary Immune Deficiency (PID) gene (RAG1, LRBA, CECR1. TACI, CARD11, CD40 and PI3K plus CASP 10) was found in 7/27 patients (26%). Four of them belonged to the group of 8 patients diagnosed with SN (50%) and 3 to that of the 6 ON subjects (50%). In 5/27(19%) patients no pathogenic gene was found. Table 1 shows clinical hematological characteristic of the 3 categories of patients.
Conclusions:In our unselected cohort of SChrNa considerable proportion (26%) of subjects bore a genetic defect that qualifies them as PID. These PID genetic defects are located in the SN and ON patients (50% each) that are clinically different from classical ELANE mutated SCN subjects because of the presence of extra-hematological signs, of markers of autoimmunity, of a diverse marrow maturation block and a frequent involvement of more than one hematopoietic lineage. While the use of NGS still leaves a not negligible proportion of SChrN without pathogenic gene, this extensive genetic diagnostic approach enables to identify a relevant portion of subjects with SChrN who indeed carry a PID genetic defect. This has important clinical implications related to specific treatment and monitoring schedules to apply to these patients. The application of the Whole ExomeSequencing technique might fill the gap of the SCHrN patient who are still gene orphan.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal